ONLINE EDITION
EUROPIN Summer School on Drug Design – Vienna
September 13 – 17, 2021
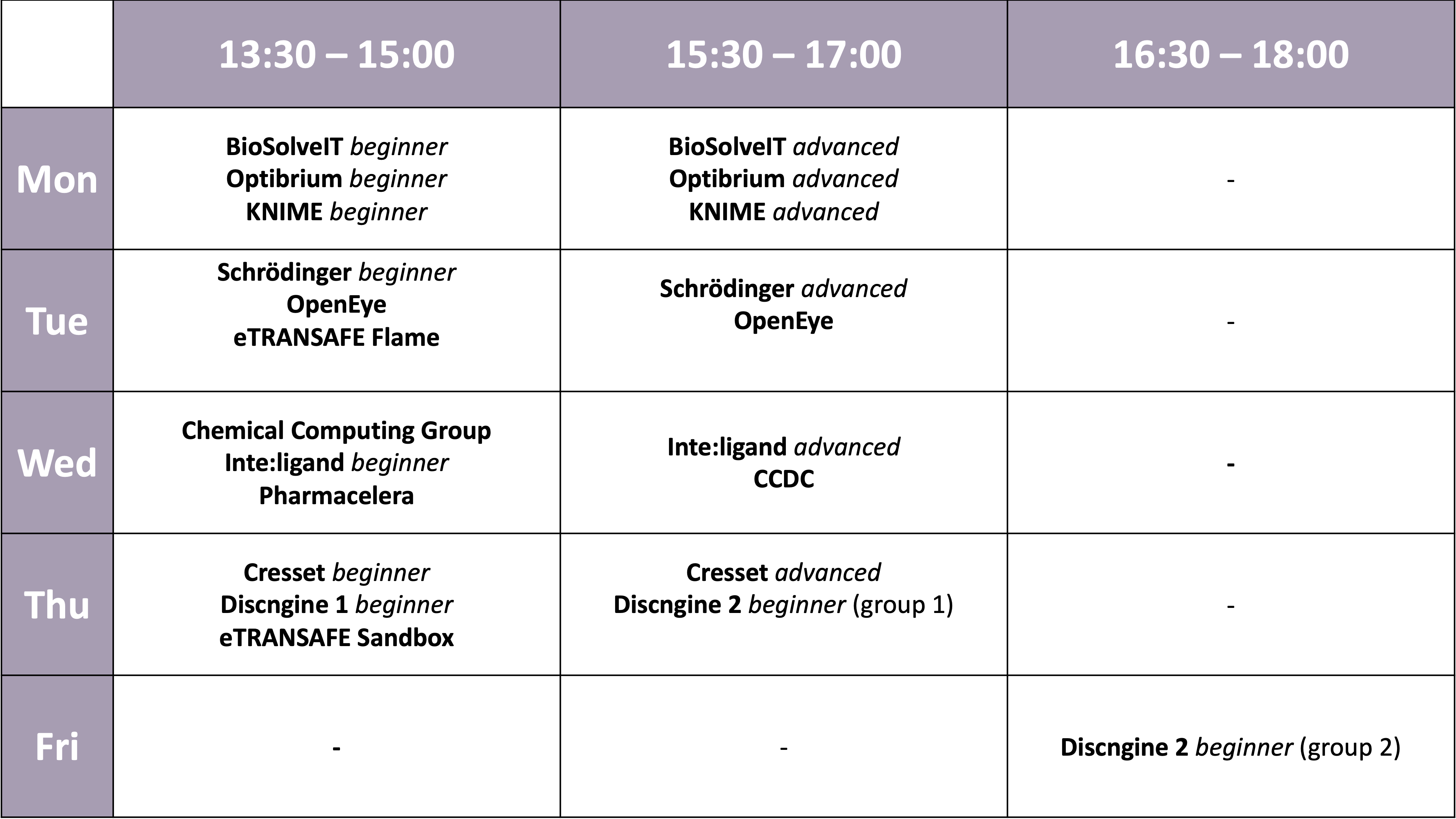
Workshops
Please click on the name in order to find more information about the workshop. Deadline for workshop registration using our Participant Area was August 31, 2021.
![]()
BioSolveIT
Interactive, Visual 3D Design with SeeSAR
Beginner’s Workshop: In this course we will start with adopting a critical view on input structures. We will then load a protein-ligand complex and grasp understanding of the binding situation: hydrogen bond determination and evaluation of their importance — in balance with desolvation, torsional analyses, and clash evaluation. You will interactively try out design ideas in the pocket and learn about their consequences: How much will unsatisfied H-bridges deteriorate your approximate ∆G? Can you reach that residue over there with an MeO-, EtO-? Of course, we will cover the basics of docking: You will place and then score ligands in binding sites. Time should permit to also exploit powerful fragment-based machineries to rescaffold and grow to explore an unoccupied subpocket while observing key parameters. All this happens in a “yin-yang” like fashion with theory and practice well balanced, while the course members can steer priorities.
Advanced Workshop: What if nothing works? The docking fails, the energy estimates are far off reality? Let us see how far we can get you using a few tricks of the trade: Use key parameters during the docking, a little prior knowledge that the algorithm can profit from, and more to be more efficient in your modeling studies. During this “advanced” part of the workshop series, we will take deeper looks at key stages in structure-based drug design: Fine-tuning the binding site definition, the setup and usage of highly definable “pharmacophores” in the pocket, mix&match approaches during merging of two ligands. In short: We will also cover the not-so-frequently used functionalities in SeeSAR, and master more complex situations with multiple proteins that can still be distilled down to swift visualization, supporting decision making. If time permits, we will also take a look at the recently implemented covalent docking features by means of an example. As in the Beginners’ part, students are welcome to steer priorities within the course.
- Limited Participation: 30 / workshop

The Cambridge Crystallographic Data Centre (CCDC)
Structure-based drug design with CSD-Discovery
In this workshop you will learn how to, given a protein-ligand complex, analyse the geometry of your ligand molecule as well as the interactions between the ligand and the protein binding site; how to simultaneously mine 3D information from both CSD and PDB structures using pharmacophore features to identify ligand modifications and scaffold hops that are tolerated in the protein binding site; how to predict the binding mode of new compounds using flexible protein-ligand docking. Applications you will use: Mogul, SuperStar, CSD-CrossMiner, GOLD.
- Limited Participation: 295 / workshop
![]()
Chemical Computing Group
SAR Analysis using MOEsaic
In this workshop we will perform a ligand-based SAR Analysis using MOEsaic on a congeneric series of COX2 inhibitors. This includes searching for and analysing Matched Molecular Pairs (MMP), creating R-Group Profiles and performing a Free-Wilson-Analysis to retrieve new compound suggestions.
- Unlimited Participation

Cresset Software
Basic workshop - Finding New Ideas for an Existing Target
In drug discovery, one of the key requirements is to design compounds which are novel. In this workshop, we will introduce you to our bioisotere replacement solution, Spark™, and show how you can replace cores from existing ligands to generate new molecules which are potentially novel. We will then go on to use features within Flare™, such as Electrostatic Complementarity™, to help prioritize the output from Spark.
Advanced workshop - Are my waters laughing?
Water molecules play an important role in ligand recognition by binding sites in biological targets. Tightly-bound (‘happy’) waters will alter the shape and electrostatic properties of the binding site and thereby influence the binding energy associated with an incoming ligand. We will use Flare™ to show where these ‘happy’ waters potentially reside in the ligand-binding site, and additionally show how the electrostatics between the protein and ligand can change if these waters are absent.
- Unlimited Participation
![]()
Discngine
Discngine 3decision® is a web-based platform that supports the ideation process and cross-team collaboration within structure-based drug discovery projects.
Workshop (beginner) 1 - How to pick the right structure for your modeling studies.
Before you start a modeling project (docking, pharmacophore modeling, molecular dynamics, etc.), you need to pick the most suitable protein structure to work on. If you pick the “wrong” structure, you can lose days (sometimes weeks) of time. Sifting through a large series of structures to assess their quality, protein conformations, ligand binding mode etc. is a tedious and sometimes tricky task. In this workshop we will show you how to use 3decision to quickly gather the PDB structures of interest, analyze their differences and create a curated data set for your structure-based drug discovery project.
Workshop (beginner) 2 - Collect medicinal chemistry ideas from cross-target exploration.
The generation of new ideas is critical to the drug design process. Thinking out-of-the-box (however cliché it sounds) is a great way of creating new starting points. In this workshop, we will show you how to use 3decision to collect ideas from other proteins and integrate them into your ligand design. Specifically, we will use the sub-pocket search to identify similar binding sites and retrieve the chemical matter that binds there. The relevant building blocks for binding will then be used to design new compounds and dock them against the primary target.
Participants at all levels are welcome to join. We offer a one-month free license on our cloud-platform after the workshop to further explore 3decision, and a dedicated Slack channel for support and assistance.
- Limited Participation: 25 / workshop
![]()
eTRANSAFE Flame
Use of Flame for the development, management, and application of predictive models.
Flame is an open-source framework for the development of predictive models. QSAR models can be generated starting from a collection of compounds annotated with biological activities. Flame implements customizable workflows that can be used to standardize the chemical structures, generate molecular descriptors, build machine learning models and validate them. Once generated, the models can be used as “predictive engines” that can be easily interchanged or deployed to predictions servers. In this workshop, the students will learn how to build QSAR models in Flame, using diverse molecular descriptors and machine learning algorithms. They will also manage these models, documenting and exporting them. The models generated by the students will be tested by predicting the properties of series of test compounds and analyzing the results. More advanced model-building techniques, like ensemble models and advanced model customization, will also be introduced in this session.
- Unlimited Participation
![]()
eTRANSAFE UNIVIE Modeling Tools
Build Machine Learning Models using Jupyter Notebook
The Jupyter Notebook is a web-based application which is used in many coding tasks since it allows an easy-to-use interface to test your code and allows a visualized interpretation of your results. In this workshop a Jupyter notebook is used to build a machine learning model in an automatic way. It is a stepwise approach, starting with the selection of meaningful data, selecting the right descriptors as well as the best suited classifier for the given dataset to obtain a well performing model. The performance of the model will be determined on statistical metrics and visualized with a confusion matrix. For this workshop no prior coding knowledge is needed.
- Unlimited Participation
![]()
Inte:Ligand
2021 Computer Aided-Drug Design and DeRisking Compounds for Neurotoxic Adverse Outcomes with LigandScout: It's not just Pharmacophores!
Inte:Ligand supports scientists worldwide with innovative software and workflows for molecule design and discovery. Join our workshops to learn modern, user friendly approaches for advanced 3D-pharmacophore modeling, virtual screening, hit triaging, target and off-target predictions, ligand profiling, docking, fragment-based approaches, cavity detection, and molecular dynamics simulations for hit finding. Learn more about derisking compounds for neurotoxic adverse outcomes using the new IL NeuroDeRisk Profiler developed as part of the NeuroDeRisk IMI-2 initiative.
Advance your molecular design projects these modern workflows.
Workshop 1: Advanced 3D-Pharmacophore Modeling and Virtual Screening Basics: Structure-Based, Ligand-Based, and Fragment-Based Approaches. Virtual Screening (VS), Library creation, and VS results analysis. LigandScout beg
Workshop 2: Hit Triaging, Target activity profiling, Neurotoxic Adverse outcomes analysis with the NeuroDeRisk Inte:Ligand Profiler; Binding affinity analysis, convenient docking, MD pharmacophores and molecule optimization with the LigandScout Apo site modeling solutions. LigandScout Adv/NeuroDeRisk IL Profiler
We will use the LigandScout Suite to explore innovative computer-aided design workflows successfully utilized by chemists and modelers in industry and academia to address major challenges they face in discovering, designing and prioritizing novel bioactive molecules.
- Limited Participation: 98 / workshop
![]()
KNIME
Build cheminformatics workflows with the open source KNIME Analytics Platform
KNIME is an open platform for the entire data science process: data access and transformation, visualization, predictive analytics, and reporting. With KNIME and its dedicated Cheminformatics Extensions you can choose from a wide range of cheminformatics functionality, which we will make use of in this workshop as well.
This year we will organize two sessions: for beginners and for advanced users of KNIME Analytics Platform.
In both sessions you will create your own interactive cheminformatics workflows that include reading, converting, visualizing and saving chemical data. In the advanced session we will offer several tasks for the group to choose from. The examples will include building components for interactive visualization, clustering molecules based on the fingerprint similarity, running a similarity search through a vendor catalog, and deploying a neural network for predictions. At the end of each session we will look at the possible solution. No prior KNIME knowledge is needed to participate in the beginner’s session. We expect though that you are a bit familiar with KNIME if you are joining the advanced session. Summing up, in addition to learning some new stuff, you will walk out with a couple of workflows that you can continue to adapt, extend and use.
- Limited Participation: 50 / workshop
![]()
OpenEye
Orion® – a cloud native platform for computer-aided drug design or how can drug-discovery in the cloud work?
Orion® is OpenEye’s reimagining of computational drug discovery and design by the cloud. It includes all of OpenEye’s software, extensive tools for data visualization and communication, useful data sources and task-oriented workflows – all in a robust, scalable, cloud environment.
Orion is a 'cloud native' platform in that all elements of Orion reside on Amazon Web Services, AWS, the world's largest on-demand computing facility. Orion uses AWS to deliver easy, scalable, maintenance-free access to hundreds, thousands, or even tens of thousands of processors, unlimited storage and archiving via reliable networks, all backed up by world-class data-security.
Orion is an open platform that allows for straightforward integration of third-party code (customer, academic, vendor). It also includes Floe, a new graphical workflow engine that makes it simple to construct scripts to take advantage of all that Orion has to offer.
In the workshop, we will show how to combine ligand- and structure-based methods in Orion, modify research protocols and analyze the results along the way.
- Unlimited Participation
![]()
Optibrium
Basic workshop - Guided Multi-Parameter Optimisation of 2D and 3D SAR
In this workshop, we will us a combination of 2D and 3D methods to understand and optimise a virtual library of Heat Shock Protein 90 (HSP90) inhibitors. We will explore the library to develop an understanding of the 3D Structure-Activity Relationship (SAR) and then use multi-parameter optimisation to further develop the absorption, distribution, metabolism and excretion (ADME) and physicochemical properties of a potent inhibitor without losing efficacy.
Advanced workshop - more information will follow
The workshop will use our StarDrop™ software and all participants will get a 1-month free trial license to use StarDrop following the workshop. For more information on StarDrop, please visit www.optibrium.com/stardrop or watch some videos of StarDrop in action at www.optibrium.com/community/videos.
- Unlimited Participation

Pharmacelera
Increasing chemical diversity using ligand-based virtual screening with PharmScreen®
Chemical diversity is a key aspect when screening virtual libraries of compounds. Hit identification must be accompanied by a scaffold diversity, exploring new chemical solutions that allow the identification of the right hit with drug-like properties while maintaining the same Mechanism of Action (MoA). For this reason, Pharmacelera has developed PharmScreen®, a ligand-based virtual screening solution that uses advanced 3D hydrophobic molecular descriptors and quantum mechanics methods. This approach describes with high accuracy the factors that determine ligand / receptor interactions. In this workshop we will describe the technology behind PharmScreen, and show how it can help you in your projects through real case examples on GPCR, CNS, …. Participants will have access to a free trial of PharmScreen.
- Limited Participation: 20 / workshop
![]()
Schrödinger
Beginner's workshop - Introduction to Small Molecule Structure Based Design
Structure-based design can be a powerful asset in any drug-discovery project, and has been a main workhorse in the pharmaceutical industry for many years. Advances in computational power, science and user interface design have made these technologies more approachable and applicable to even larger datasets than before. The workshop will introduce the basic concepts of structure-based drug design, and provide attendees with a general understanding of how structure-based virtual screening works. Participants will get the opportunity to gain some hands-on experience with the Schrödinger software, and apply what they have learned in a tailored case study. Specific topics covered will include protein and ligand preparation, setting up ligand docking calculations for virtual screening, visualization of results, and interactive structure based design using LigandDesigner.
Advanced workshop - Biologics Modeling: Antibody and Protein Design
Biologics are one of the most important classes of therapeutics, be it for vaccine design, the design of therapeutic antibodies or the design of newer modalities such as antibody-drug-conjugates (ADCs). Despite this, the application of rational design strategies remains challenging for these modalities. Recently, in silico technologies have emerged as valuable tools for designing and optimizing biologics. In this workshop, participants will learn how reliable computational solutions can be applied during research and development to direct experimental effort, with the aim of reducing costs and timelines. Participants will have the opportunity to gain hands-on experience on typical rational design techniques such as protein stability engineering, antibody optimization and aggregation prediction.
- Unlimited Participation
Contact us
If you have any questions regarding the EUROPIN Summer School on Drug Design, please don’t hesitate to contact us at summerschool@europin.at
EUROPIN – a structured, highly interconnected training through research PhD program on the efficient and innovative use of information technologies and computational approaches in the drug discovery, design and development processes.
Organised by
Pharmacoinformatics Research Group
Following a holistic pharmacoinformatic approach we combine structural modeling of proteins, structure-based drug design, chemometric and in silico chemogenomic methods, statistical modeling and machine learning approaches to develop predictive computational systems for transporters and ion channels.
The validation and optimisation of the obtained in silico models by strong links to experimental groups is an integral part of these activities.