ONLINE EDITION
EUROPIN Summer School on Drug Design – Vienna
September 13 – 17, 2021
Program
Status: final
Please find the agenda of the Summer School on Drug Design 2021 in Vienna underneath or download the latest version here. All times on this program are in the local time zone (CEST - Central European Summer Time)! Please see Timezone Information for guidance.
Since we will unfortunately not be able to personally welcome our participants in Vienna this year, there will be a virtual city tour through Vienna as a special extra. The program will of course also contain breaks and socializing events - read more at Organisation.
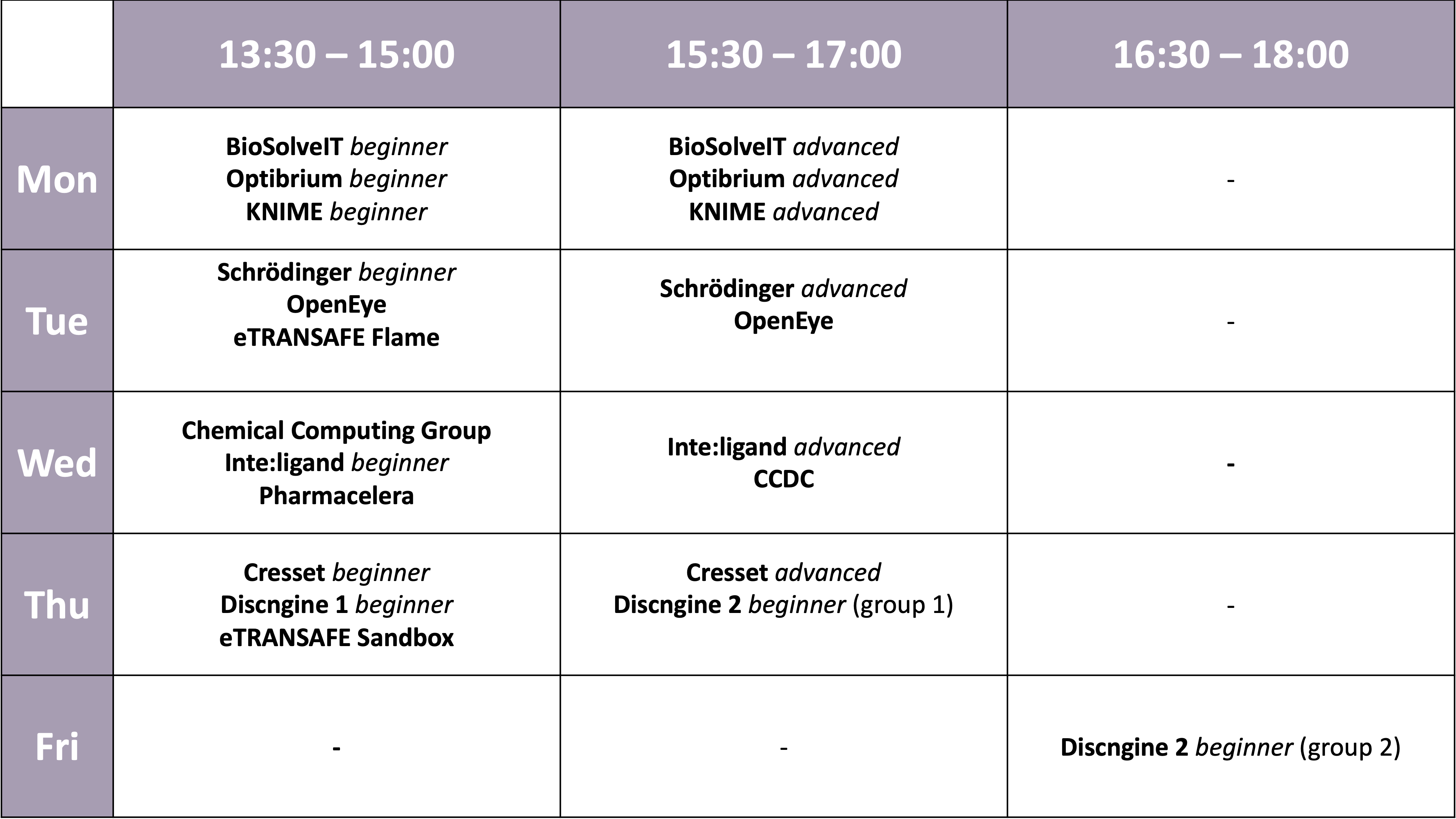
| 08:15 - 08:30 | Welcome | |
| 08:30 - 09:00 | Thierry Langer – University of Vienna | Adventures in Computer-Assisted Molecular Design |
| 09:00 - 09:30 | Stefan Boresch – University of Vienna | Setting up MD Simulations of Biomolecules |
| 09:30 - 10:00 | Chris Oostenbrink – BOKU Vienna | Applications of free energy calculations from molecular dynamics simulations |
| 10:00 - 10:30 | Coffee Break | |
| 10:30 - 11:00 | Andrea Cavalli – University of Bologna | Dynamic docking and free energy estimation approaches to drug discovery |
| 11:00 - 11:30 | Daria Goldmann – KNIME | Predicting bioactivity in KNIME Analytics Platform |
| 11:30 - 12:00 | Marcus Gastreich – BioSolveIT | Navigating Septillion-Sized Chemical Spaces |
| 12:00 - 12:30 | Matt Segall – Optibrium | Multi-parameter Optimisation in Drug Discovery: Targeting compounds with a high chance of success |
| 12:30 - 13:30 | Lunch Break | |
| 13:30 - 15:00 | Workshops | Simultaneously: BioSolveIT beginners / Optibrium beginners / KNIME beginners |
| 15:00 - 15:30 | Coffee Break | |
| 15:30 - 17:00 | Workshops | Simultaneously: BioSolveIT advanced / Optibrium advanced / KNIME advanced |
| 17:00 - 17:30 | Break | |
| 17:30 | Special | Virtual City Tour through Vienna |
| 08:30 - 09:00 | Gerhard Ecker – University of Vienna | Integrated approaches for toxicity prediction |
| 09:00 - 09:30 | Andrea Volkamer – Charité Universitätsmedizin Berlin | In silico Tools to Support Risk Assessment of Small Molecules |
| 09:30 - 10:00 | Johannes Kirchmair – University of Vienna | In silico prediction of drug metabolism |
| 10:00 - 10:30 | Coffee Break | |
| 10:30 - 11:00 | Manuel Pastor – Universitat Pompeu Fabra | Application of knowledge-based computational methods in Toxicology |
| 11:00 - 11:30 | Philipp Jäger – Boehringer-Ingelheim | Integrative data approaches for smart PROTAC designs |
| 11:30 - 12:00 | Gunther Stahl – OpenEye | Virtual Screening - From small to LARGE scale - from local to the cloud |
| 12:00 - 12:30 | Stephan Ehrlich – Schrödinger | Structure-based screening of large chemical libraries |
| 12:30 - 13:30 | Lunch Break | |
| 13:30 - 15:00 | Workshops | Simultaneously: Schrödinger beginners / OpenEye / eTRANSAFE Flame |
| 15:00 - 15:30 | Coffee Break | |
| 15:30 - 17:00 | Workshops | Simultaneously: Schrödinger advanced / OpenEye |
| 17:00 - 17:30 | Break | |
| 17:30 - 19:00 | Career Session | Panel discussion with: Margot Ernst (Medical University of Vienna), Daria Goldmann (KNIME), Alexander Hillisch (Bayer AG), Martin Kotev (Evotec), Floriane Montanari (Bayer AG), Doris Schütz (IRIC - University of Montreal), Alžběta Türková (Uppsala University); Moderation: Gerhard Ecker (University of Vienna) |
| 08:30 - 09:00 | Margot Ernst – Medical University of Vienna | Homology modeling in the gray zone of low sequence similarity |
| 09:00 - 09:30 | Wolfgang Sippl – Martin Luther University of Halle-Wittenberg | Structure based design of selective ligands for epigenetic targets |
| 09:30 - 10:00 | Gerhard Wolber – Freie Universität Berlin | In silico pharmacology: Pyrod and dynophores as powerful computational microscopes to decode receptor function |
| 10:00 - 10:30 | Coffee Break | |
| 10:30 - 11:00 | Barbara Zdrazil – University of Vienna | Deciphering the molecular basis of ligand-recognition and selectivity in hepatic Organic Anion Transporting Polypeptides |
| 11:00 - 11:30 | F. Javier Luque – University of Barcelona / Pharmacelera | From simple solvation models to applications in target druggability and screening of drug-like compounds |
| 11:30 - 12:00 | Sharon Bryant – Inte:Ligand | De-Risking Compound Structures for Neurotoxicity: The NeuroDeRisk Inte:Ligand Profiler |
| 12:00 - 12:30 | Andrew Henry – Chemical Computing Group | MOEsaic: Guiding Multi-Parameter Optimization in Ligand-Based Design |
| 12:30 - 13:30 | Lunch Break | |
| 13:30 - 15:00 | Workshops | Simultaneously: CCG / LigandScout beginner / Pharmacelera |
| 15:00 - 15:30 | Coffee Break | |
| 15:30 - 17:00 | Workshops | Simultaneously: CCDC / LigandScout advanced |
| 17:00 - 17:30 | Break | |
| 17:30 | Poster Session 1 | Posters 1-36 |
| 08:30 - 09:00 | Dušanka Janežič – University of Primorska | Protein Binding Sites Tools for Innovative Drug Design |
| 09:00 - 09:30 | Alexander Hillisch – Bayer AG | Design and Preclinical Characterization Program Towards BAY 2433334, an Oral Factor XIa Inhibitor for the Prevention and Treatment of Thromboembolic Disorders |
| 09:30 - 10:00 | Claire Colas – University of Vienna | Structure based ligand discovery methods applied to Solute Carrier Transporters |
| 10:00 - 10:30 | Coffee Break | |
| 10:30 - 11:00 | Floriane Montanari – Bayer AG | An introduction to explainable AI for small molecules |
| 11:00 - 11:30 | Daniela Digles – University of Vienna | Analysing Solute Carrier (SLC) substrates with KNIME |
| 11:30 - 12:00 | Stuart Firth-Clark – Cresset | Using Spark™ and Flare™ to design and prioritize novel molecules in a drug design project |
| 12:00 - 12:30 | Lorena Zara – Discngine | Learn how to navigate the vast and rich protein structural space with 3decision |
| 12:30 - 13:30 | Lunch Break | |
| 13:30 - 15:00 | Workshops | Simultaneously: Cresset beginner / Discngine beginner 1 / eTRANSAFE Sandbox |
| 15:00 - 15:30 | Coffee Break | |
| 15:30 - 17:00 | Workshops | Simultaneously: Cresset advanced / Discngine beginner 2 (group 1) |
| 17:00 - 17:30 | Break | |
| 17:30 | Poster Session 2 | Posters 37-72 |
| 08:30 - 09:00 | Abhik Mukhopadhyay – CCDC | Ligand based virtual screening in Drug discovery |
| 09:00 - 09:30 | Klaus-Jürgen Schleifer – BASF | Learning from Ligands |
| 09:30 - 10:00 | Gerhard Hessler – Sanofi | Computational Design of macrocyclic compounds |
| 10:00 - 10:30 | Coffee Break | |
| 10:30 - 10:45 | Oliver Wieder – University of Vienna | Improved lipophilicity and aqueous solubility prediction with composite graph neural networks. |
| 10:45 - 11:00 | Theresa Noonan – Freie Universität Berlin (EUROPIN Student) | Inhibiting Bacterial Ribosomal Assembly as a Novel Antibiotic Approach |
| 11:00 - 11:15 | Theres Friesacher – University of Vienna | Molecular Dynamic Simulations of Ion Channels: Investigating a Rare Disease Mutation through the Computational Microscope |
| 11:15 - 11:30 | Dominique Sydow - Charité Universitätsmedizin Berlin | KiSSim: Subpocket-Enhanced Kinase Similarity Assessment for Off-Target Prediction |
| 11:30 - 11:45 | Barbara Füzi – University of Vienna (EUROPIN Student) | Pathway and network studies of toxic compounds |
| 11:45 - 12:00 | Doha Naga – Hofmann La Roche / University of Vienna | Automated machine learning in drug discovery |
| 12:00 - 12:15 | Stefan Michael Kohlbacher – University of Vienna | QPhAR: Quantitative Pharmacophore Activity Relationship |
| 12:15 - 12:30 | Christian Permann – University of Vienna | Greedy 3-Point Search (G3PS) - A novel algorithm for pharmacophore alignment |
| 12:30 - 13:30 | Closing & Lunch Break | |
| 13:30 - 16:30 | EUROPIN Presentations | from EUROPIN students and applicants (see agenda below) |
| 16:30 - 18:00 | Workshops | Discngine beginner 2 (group 2) |
| 13:30 - 13:45 | Szymon Pach – Freie Universität Berlin | Tracing interactions of SARS Cov2 spike protein in complex with animal ACE2 ortholgs |
| 13:45 - 14:00 | Nguyen Trung Ngoc – Freie Universität Berlin | ACE2-variants indicate potential SARS-CoV-2 susceptibility in animals: data analysis |
| 14:00 - 14:15 | Pablo Rodríguez Belenguer – Pompeu Fabra University | Combining machine learning models for improving their predictive quality and usefulness in biomedical applications |
| 14:15 - 14:30 | Kristina Puls – Freie Universität Berlin | Mechanistic investigation and development of tailored modulators of the Kappa opioid receptor |
| 14:30 - 14:45 | Nejra Granulo – University of Vienna | The macrocyclic landscape of SLCs |
| 14:45 - 15:00 | Alexander Wolf – Freie Universität Berlin | In silico modelling of human Cytochrome P450 enzymes |
| 15:00 - 15:15 | Break | |
| 15:15 - 15:30 | Aljoša Smajić – University of Vienna | Decentralized machine learning approaches for toxicity predictions |
| 15:30 - 15:45 | Francesca Galvani – University of Parma | Multiscale simulations for the design of new drug-like compounds |
| 15:45 - 16:00 | Jiahui Huang – University of Vienna | Structure landscape analysis and functional mapping of mutations on SLC transporters |
| 16:00 - 16:15 | Gian Marco Elisi – University of Parma | Free-energy simulations on melatonin receptors ligands |
| 16:15 - 16:30 | Closing & Break | |
| 16:30 - 17:30 | EUROPIN Board Meeting | Closed Session |
Contact us
If you have any questions regarding the EUROPIN Summer School on Drug Design, please don’t hesitate to contact us at summerschool@europin.at
EUROPIN – a structured, highly interconnected training through research PhD program on the efficient and innovative use of information technologies and computational approaches in the drug discovery, design and development processes.
Organised by
Pharmacoinformatics Research Group
Following a holistic pharmacoinformatic approach we combine structural modeling of proteins, structure-based drug design, chemometric and in silico chemogenomic methods, statistical modeling and machine learning approaches to develop predictive computational systems for transporters and ion channels.
The validation and optimisation of the obtained in silico models by strong links to experimental groups is an integral part of these activities.